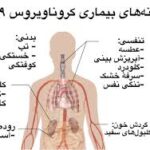
بیماری لکودیستروفی چه تاًثیری روی رشد فیزیکی فرددارد ؟

زندگی با دیستروفی عضلانی (MD) می تواند فوق العاده چالش برانگیز باشد. یکی از معمول ترین سوالاتی که بیماران می پرسند این است که طول عمر بیماران مبتلا به دیستروفی عضلانی چقدر است. اغلب اوقات، داستان هایی از نگرانی والدین درباره آینده فرزندانشان می شنویم. این والدین در وضعیت عدم اطمینان زندگی می کنند. این قابل درک است که بخواهید از چشم انداز زندگی بیماران مبتلا به MD در آینده مطلع شوید.
یک مسئله اصلی برای کسانی که دیستروفی عضلانی دارند، طول عمر است. MD می تواند زندگی روزمره را دشوار کند. علاوه بر این، زندگی این افراد می تواند از نظر جسمی و روانی ناتوان کننده باشد. اما درک امید به زندگی در بزرگسالی برای برنامه ریزی آینده ضروری است. امید به زندگی از بیماری به بیمار دیگر به دلیل انواع مختلف دیستروفی عضلانی و درجات شدت آن متفاوت است.
در این پست به بررسی اشکال مختلف دیستروفی عضلانی می پردازیم. ما طول عمر معمول افرادی را که به اشکال مختلف این بیماری مبتلا هستند، کشف خواهیم کرد.
ما همچنین کشف خواهیم کرد که چگونه پیشرفت پزشکی می تواند طول عمر بیماران مبتلا به دیستروفی عضلانی (MD) را طولانی تر کند. سپس بیماران می توانند تصمیم بگیرند که چگونه به روشی آگاهانه به درمان بیماری خود نزدیک شوند.
دیستروفی یا دُشپروردگی ماهیچهای دوشن (به انگلیسی: Duchenne muscular dystrophy) (به اختصار DMD)، شایعترین و شدیدترین نوع از بیماریهای دیستروفی ماهیچهای از گونه (X پیوند یافته) و ژنتیکی است. از عوارض اصلی این بیماری تحلیل و نابودی ماهیچههای ارادی (که در کنترل بدن نقش حیاتی دارند) است که در مواقع شدید نهایتاً منجر به عدم توانایی در راه رفتن ،۹۶٪ معلولیت، مشکلات تنفسی و مرگ میشود.
فراوانی دوشن ۱ در ۳۵۰۰ کودک پسر است. این بیماری در اثر تغییرات و نقض در ژن (DMD) واقع در کروموزوم X ایجاد میشود. این ژن مسؤل ساخت پروتئین دیستروفین که مادهای ضروری برای حفظ و ثبات ماهیچهها است میباشد و در غشاء سلولی قرار دارد. بهطور کلی بانوان دارای کروموزوم X ناقص و حامل این بیماری هستند و میتوانند این بیماری را به نیمی از فرزندان پسر خود منتقل کنند. ناقص بودن یکی از کروموزوم X در بانوان غالباً ارثی است یا ممکن است در اثر جهشهای ژنتیکی باشد. بهطور کلی علایم بیماری در ۶ سالگی ظاهر میشود گرچه در دوران نوزادی هم نمایان است. نوزاد در موقع تولد کاملاً سالم به نظر میرسد، ولی هنگامی که شروع به حرکت میکند، حتی زمانی که با چهار دست و پا یا سینه خیز مینماید حرکت او کندتر از همسالان خود است و زود به زود خسته میشود. هنگامی که به راه میافتد ضعف ماهیچهای به تدریج واضح تر میشود. معمولاً والدین نسبت به نحوه راه رفتن غیرطبیعی یا ۰اشکال در برخاستن فرزندشان از روی زمین، راحت زمینخوردن، اشکال در بالا رفتن از پلهها و عدم توانایی دویدن یا انجام حرکات ورزشی در مقایسه با همسالانشان نگران میشوند. بزرگ شدن غیرطبیعی ماهیچههای ساق پا و ضعف خفیف تا متوسط ماهیچههای ابتدای اندام تحتانی، به صورت راه رفتن اردک وار و عدم توانایی در برخاستن راحت از روی زمین تظاهر میکنند. این کودکان بهطور مشخص برای برخاستن از حالت نشسته از دستها و بدنشان کمک میگیرند که به نشانه گاور Gowers’s sign معروف است. تحلیل ماهیچهای به صورتی است که در ۸ سالگی کاملاً ضعف ماهیچهها بارز میشود. این تحلیل ماهیچهای ابتدا از پاها و لگن شروع، باعث کاهش حجم و نابودی ماهیچههای این ناحیه میشود و نهایتاً دستها، بازوها و شانه را نیز در بر میگیرد که موجب افتادگی شانه میشود. در ۱۰ سالگی بیمار توانایی راه رفتن را از دست میدهد و به ویلچر نیاز پیدا میکند. از آنجایی که دو سوم از وزن بدن از ماهیچهها تشکیل شدهاست، با نابودی ماهیچهها وزن بدن به شدت کاهش مییابد و به یک سوم از کل آن میرسد. در بیشتر مواقع پیشرفت بیماری ادامه مییابد و به علت رشد غیرطبیعی استخوانهای ستون فقرات، ستون فقرات به صورت حرف S انحنا پیدا میکند و قفسه سینه اندکی تغییر شکل میدهد که سبب جمع شدگی بدن میشود. پیشرفت بیماری در مواقع شدید نهایتاً با مشکلات تنفسی و مرگ همراه خواهد بود، طول عمر متوسط برای بیماران معمولاً بین ۲۰ تا ۳۰ سال است.
شیوع
شیوع بیماری دوشن ۱ در ۳۵۰۰ کودک پسر است. به این دلیل این بیماری فقط در جنس مذکر بروز میکند که جنس مذکر فقط یک کروموزوم ایکس دارد. جنس مؤنث به این بیماری مبتلا نمیشود زیرا که در بدن آنها دو کروموزوم ایکس وجود دارد و دوشن فقط در اثر اختلال یکی از کروموزومهای ایکس ایجاد میشود بنابراین آن یکی کروموزوم سالم میماند و نهایتاً فقط پسران به این بیماری مبتلا میشوند.
نام بیماری
نام بیماری دیستروفی ماهیچهای دوشن برگرفته از نام یک پزشک مختصص مغز و اعصاب فرانسوی به نام گیوم بنیامین اماند دوشن است که اولین بار در سال ۱۸۶۱ شرح بیماری دوشن را در مجلات پزشکی آن زمان منتشر کرد.
عامل بیماری
بیماری دیستروفی ماهیچهای دوشن در اثر نقض در کروموزوم ایکس و فقدان پروتئین دیستروفین ایجاد میشود. ژن مسئول ساخت این پروتئین (دی ام دی) است و بر روی کروموزوم ایکس قرار دارد. پروتئین دیستروفین اطراف سلولهای ماهیچهای برای محافظت ساختمان ماهیچه ساخته میشوند و مانع از خروج عناصر داخل سلول ماهیچهای به فضای خارج از سلول میشود. بدون دیستروفین سلول ماهیچهای قابل نفوذ خواهد بود و مواد بافت خارج سلولی وارد سلول ماهیچه شده و باعث تخریب و مرگ ماهیچه خواهد شد و در نهایت بافت چربی جای ماهچه را میگیرد. علت عدم تولید پروتئین دیستروفین در ژن دی ام دی هنوز بهطور قطع مشخص نیست ولی این عدم تولید ناشی از مسدود شدن مجاری تولید پروتئین دیسترفین میباشد. پروتئین دیستروفین در بدن افراد طبیعی کمتر از ۰٫۰۰۲٪ میلیگرم از کل پروتئین بافت ماهیچهای است.
نشانهها و عوارض
مهمترین نشانه بیماری دیستروفی ماهیچهای دوشن اختلال در ماهیچه هاست که موجب مشکل در راه رفتن، ضعف ماهیچههای اندام تحتانی و لگن و برزگ شدن غیرطبیعی ماهیچههای ساق پا میشود. همچنین ضعف ماهیچهای در دستها، بازو و شانه نیز بروز میکند. نشانهها معمولاً تا قبل از ۶ سالگی ظاهر میشود، هرچند در دوران نوزادی هم نمایان است. نهایتاً پیشرفت این بیماری که موجب نابودی ماهیچهها میشود با عدم توانایی راه رفتن، ۹۶٪ معلولیت، انحنا در ستون فقرات و قفسه سینه، مشکلات تنفسی و مرگ خاتمه مییابد. سایر نشانهها و عوارض اصلی دوشن به شرح زیر است:
- ناهنجاری در راه رفتن و قدم برداشتن (بیماران معمولاً روی قسمت جلویی کف پا و سر پنجه، راه میروند که ناشی از ضعف و کاهش قدرت در عضلات بازکننده (یا همان اکستنسور) زانوها میباشد)
- خستگی زود هنگام و درد در ماهیجههای ساق پا پس از مدت کوتاهی پیادهروی
- زمینخوردن مکرر
- مشکل در دویدن، جستن و پریدن
- مشکل در بالا رفتن و پایین آمدن از پله
- مشکل در انجام حرکات ورزشی
- بزرگ شدن غیرطبیعی ماهیجههای ساق پا به دلیل تخریب و تحلیل سلولهای عضلانی و بافت ماهیچهای و جایگزین شدن بافت چربی و بافت فیبری به جای آن
- کوتاه شدن تاندهای آشیل پا
- عدم توانایی در برخاستن راحت از روی زمین و کمک از دستها برای بلند شدن (نشانه گاور) Gowers’ sign
- تحلیل ماهیجههای بازو و شانه
- از دست دادن توانایی راه رفتن
- ۹۶٪ معلولیت (بهطوریکه فرد بیمار به تنهایی قادر به زندگی نیست)
- ضعیف شدن دستگاه گوارش و اشکال در دفع
- انحنا در ستون فقرات و قفسه سینه در مواقع شدید
- مشکلات تنفسی در مواقع شدید
- تحلیل ماهیچههای قلب در مواقع شدید
- مرگ در مواقع شدید
نشانههای نهفته و موجود در خون
- بالا بودن میزان آنزیم ماهیچهای کراتین کیناز «Creatine kinase (CPK-MM)» در خون
- تست ژنتیک نقص در کرموزوم ایکس را افشا میکند
- الکترومیوگرافی از بافت عضلانی نشان میدهد که تحلیل عضلات ناشی از با نفوذ شدن بافت عضلانی در اثر نقص در تولید پروتئین دیستروفین است نه در اثر اعصاب.
- نمونه برداری از بافت ماهیچهای فقدان پروتئین دیستروفین را تأیید میکند.
تست DNA
ژن دیستروفین مرکب از ۷۹ اگزون است و تجزیه و تحلیل تست DNA معمولاً نوع خاصی از جهش در اگزون یا اگزونهای فرد مبتلا را مشخص میکند. تست DNA در اکثر موارد ابتلای فرد به بیماری را تأیید میکند.
نمونه برداری از ماهیچه
اگر تست DNA نتواند جهش در اگزونها را پیدا کند، ممکن است از ماهیچه نمونه برداری شود. نمونه کوچکی از ماهیچه استخراج شده (معمولاً با چاقوی کوچک جراحی به جای سوزن سرنگ) وضعیت فقدان کامل پروتئین دیستروفین را نشان میدهد. در طول چند سال گذشته تست DNA در زمینه شناسایی جهشهایی زیادی در ژنها که سبب ایجاد بیماریهای مختلف میشود پیشرفت داشتهاست و نمونه برداری از ماهیچه غالباً تنها بیماری دوشن را تأیید نمیکند.
آزمایش پیش از تولد
اگر یکی یا هردو والدین حامل بیماری خاصی باشد، مبتلا شدن فرزند به دنیا نیامده آنها به آن بیماری ریسک است. آزمایش پیش از تولد برای پیدا کردن جنین مبتلا به دوشن در دوران بارداری انجام میشود، این آزمایشها تنها برای برخی از اختلالات عصبی و ماهیچهای کاربرد دارد. انواع آزمایشهای پیش از تولد میتواند بعد از حدود ۱۱ هفته پس از بارداری به اجرا دربیاید. نمونه برداری از برآمدگی مویی پرده بیرونی جنین (CVS) در هفته ۱۱ تا ۱۴ بارداری انجام و تست پرده آمنیون مایع بعد از هفته ۱۵ بارداری انجام میشود، در حالی که نمونه خون جنین میتواند حدوداً در هفته ۱۸ بارداری صورت گیرد. زنان یا زوجها باید دقت نظر داشته باشند که پس از انجام آزمایش در مورد آن با مشاور ژنتیک به بحث بنشیند. در صورتی که آزمایش پیش از تولد ابتلا جنین به بیماری دوشن را مشخص کند دستور سقط جنین صادر میشود تا از تولد نوزاد بیمار جلوگیری شود.
افسردگی بیماران
از آنجا که بیماری دوشن یک بیماری بسیار حاد است و در مدت کوتاهی زندگی بیماران و خانوادههای آنان را دگرگون میکند، عمدتاً مبتلایان به این بیماری را در کودکان، نوجوانان و جوانان تشکیل میدهند، به علت عدم پذیرش معلولیت از سوی مبتلایان و به خصوص جامعه که برای بیماران هیچ گونه تمهیدات، مناسبسازی و برنامهای انجام نداده و عدم فرهنگسازی که مردم با ترحم به بیماران نگاه میکنند. موارد زیادی از افسردگی میان جوانان مبتلا به این بیماری وجود دارد. همچنین دیستروفی ماهیچهای دوشن به دلیل نوع پیشرونده بودن بیماری، نهایتاً منجر به معلولیت شخص میشود، این موضوع میتواند عوارض روحی و روانی را در مبتلایان و خانوادههای آنها را به دنبال داشته باشد. به دلیل این مسئله حتی چند مورد خودکشی مادران بیماران گزارش شدهاست.
درمان
لوکودیستروفی TUBB4a یک بیماری ژنتیکی غیرقابل درمان است که به سیستم عصبی مرکزی آسیب میرساند و میتواند برای نوزادان و کودکان خردسال کشنده باشد. TUBB4a به عنوان کمبود ماده سفید همراه با آتروفی عقدههای قاعدهای و مخچه، یک نوع رایج لوکودیستروفی، نشان داده میشود. در ابتدا، دانشمندان بر این باور بودند که این بیماری تنها ۲۰۰ نفر را در سراسر جهان تحت تاثیر قرار داده است. با این حال، تحقیقات اخیر نشان میدهند که این تعداد ممکن است بسیار بیشتر از آن چه قبلا تصور میشد، باشد. لوکودیستروفی از هر ۷۶۶۳ تولد یک نفر را تحت تاثیر قرار میدهد. با حدود ۱۴۰ میلیون کودک متولد شده در جهان در سال ۲۰۲۱، این بدان معنی است که بیش از ۱۸۰۰۰ کودک ممکن است تنها در سال گذشته مبتلا به لوکودیستروفی و حدود ۱۶۵۰ کودک مبتلا به TUBB4a بوده باشند. دکتر دان ویلیامز، مدیر عامل و یکی از بنیانگذاران شرکت بیوتکنولوژی SynaptixBio، که هدف آن توسعه اولین درمان در جهان برای این بیماری ناتوان کننده است، گفت: تعداد واقعی بیماران TUBB4aممکن است حتی بیشتر باشد.یکی از مشکلات اصلی در تشخیص بیماری مانند TUBB 4a این است که تشخیص آن بسیار دشوار است. برای شناسایی صحیح آن نیاز به توالی یابی ژنوم و اسکنهای MRI گسترده است. لوکودیستروفی TUBB4a برای اولین بار در سال ۲۰۱۴ شناسایی شد و در حال حاضر ۹ ٪ از یک گروه از حدود ۵۰ اختلال عصبی نادر را که به عنوان لوکودیستروفی شناخته میشوند، تشکیل میدهد. ضروری است که درک ما از این بیماری به سرعت بهبود یابد. در حالی که هنوز یک بیماری نسبتاً نادر است، واضح است که نیاز به یافتن درمانی بیش از آن چه تخمینهای اولیه ممکن است تصور شود، وجود دارد. این وضعیت به دلیل جهش در ژن TUBB4a ایجاد میشود که سیگنالهایی را که بین نورونهای مغز منتقل میشود، مختل میکند. این اختلال عصبی میتواند باعث شود بیماران در راه رفتن، نشستن و بلع مشکل داشته باشند. آنها میتوانند تشنج، اسپاسم عضلانی، مشکلات شنوایی و گفتار و حرکات غیرقابل کنترل اندام را ایجاد کنند. دکتر مایکل تنگ، یکی از بنیانگذاران و رئیس علوم SynaptixBio، که دختر ۱۱ ساله اش، سوفیا، مبتلا به TUBB4a شدید معروف به H-ABC را است؛ خواستار دسترسی بیشتر به آزمایشهای تشخیصی برای بیماران بالقوه شد . SynaptixBio که سال گذشته راه اندازی شد، در حال حاضر روی یک درمان امیدوارکننده کار میکند که امیدواریم در نحوه درمان TUBB4a را تحول ایجاد کند.دانشمندان امیدوارند که درمان با آنتاگونیستهای نوکلئوتیدی (ASOs) که قبلا برای درمان بیماریهایی مانند دیستروفی عضلانی دوشن و آتروفی عضلانی نخاعی استفاده میشد، به طور قابل توجهی کیفیت و طول عمر بیماران لوکودیستروفی را افزایش دهد.
تاکنون هیچ درمانی برای دیستروفی ماهیچه ای دوشن یافت نشده است اگرچه اخیراً تحقیقات نشان میدهد به وسیله سلولهای بنیادی میتوان بافت ماهیچه های سالم را جایگزین بافت ماهیچهای آسیب دیده کرد. تنها با فیزیوتراپی و کاردرمانی میتوان از انحراف ستون فقرات و قفسه سینه که موجب جمع شدگی بدن میشود جلوگیری کرد.
درمان کورتیکواستروئیدی[۱] با عوارض جانبی مختلفی از جمله افزایش وزن ناخواسته، تاخیر در رشد، تاخیر در شروع بلوغ، ضعف استخوانی و نارسایی آدرنال همراه است. در سال های اخیر، مناسب بودن درمانهای ژنی و آنتی سنس[۲] برای درمان DMD با رویکردهای مختلف مورد بررسی قرار گرفته است و چهار داروی کاندید ، تاییدیه FDA را دریافت کرده اند و اکنون برای درمان زیر مجموعه خاصی از بیماران DMD در بازار موجود هستند.
پیش آگهی
دیستروفی عضلانی دوشن یک بیماری پیشرونده نادر است که در نهایت تمام عضلات ارادی را تحت تاثیر قرار می دهد و عضلات قلب و تنفس را در مراحل انتهایی درگیر می کند. امید به زندگی در حدود ۲۵-۲۶ سال تخمین زده می شود[۳][۴] ولی در افراد مختلف متفاوت است. با مراقبت های پزشکی کامل مردان مبتلا به این بیماری تا ۳۰ سالگی هم عمر می کنند.[۵] دیوید هتچ از maine در ایال
نادیده انگاری اگزونی
کارآزمایی بر روی “AVI ۴۶۵۸” که یک فراورده آزمایشگاهی برای نادیده انگاشتن اگزونی در دیستروفی ماهیچهای دوشن است موجب تولید دیستروفین شده و بیخطر به نظر میرسد. “AVI ۴۶۵۸” یک داروی آزمایشگاهی است که توسط شرکت “AVI BioPharma” برای نادیده انگاشتن اگزون ۵۱ ژن دیستروفین تولید شدهاست. نادیده انگاشتن اگزون ۵۱ توان بالقوه برای تولید پروتئین دیستروفین را در زیر مجموعهای از افراد مبتلا به دیستروفی ماهیچهای دوشن را دارد. هنگامی که دارو به شکل داخل وریدی داده شد، بیخطر به نظر رسید و سبب افزایش تولید دیستروفین در برخی از داوطلبان گردید. ۲۴ پسر بین ۵ تا ۱۵ سال دارای جهش دیستروفین در ناحیهٔ اگزون ۵۱ داروی “AVI ۴۶۵۸” را به شکل هفتگی به مدت ۱۲ هفته در یکی از شش مقیاس دوزاژ دریافت کردند. هر گروه نسبت به گروه قبلی مقدار بیشتری از دارو را دریافت کد. دارو بی هیچ رویداد ناگواری تحمل گردید، هر چند در یکی از پسرها ضعف عضله قلبی (کاردیو مایو پاتی) تشدید شد و از کارآزمایی کنار گذاشته شد. اینکه آیا دارو سبب بروز این مشکل شد یا خیر مشخص نگردید، شرکت “AVI ۴۶۵۸” قصد ادامه آزمایش دارو را دارد.
سلولهای بنیادی
محققان «بیمارستان اطفال پیتسبورگ» دسته منحصربهفردی از سلولهای بنیادی بزرگسال را شناسایی کردهاند که میتواند بیماریهایی نظیر حمله قلبی و دیستروفی ماهیچهای دوشن را درمان کند. دانشمندان سلولهای بنیادی «میوآندوتلیال» myoendothelial را جداسازی و مشخص کردند، این سلولها با استفاده از فناوریهای جداسازی سلول به راحتی جدا میشوند، به سرعت تکثیر مییابند و میتوانند در محیط آزمایشگاه به سلولهای ماهیچه ای، استخوان و غضروف تمایز پیدا کنند. کشف این دسته سلولهای بنیادی در یک منبع انسانی موفقیت بزرگی بهشمار میرود، زیرا به کاربرد بالینی این درمان بسیار نزدیکتر میشود. به گفته دانشمندان، پیوند هزار سلول میوآندوتلیال به ماهیچه آسیب دیده موشهایی که دچار نقص ایمنی بودند بهطور متوسط ۸۹ لیف عضلانی تولید کرد.